

Unterschied zwischen Amyloid und Prion
- 1055
- 288
- Tina Gürbig
Amyloide und Prionen sind beide gleichbedeutend mit neurodegenerativen Störungen, Störungen, die Degeneration innerhalb des Gehirns verursachen. Amyloide sind nicht hirnspezifisch, da sie in Organen im Körper Störungen verursachen können. In Bezug auf die Neurologie sind die menschlichen neurodegenerativen Störungen, für die Amyloide und Prionen direkt oder indirekt verantwortlich sind, solche wie Alzheimer-Krankheit, Parkinson-Krankheit und Creutzfeldt-Jakob-Krankheit. Das Verständnis der Funktionsweise innerhalb von Amyloiden ist ein wichtiger Aspekt beim Verständnis von Prionen und Neurodegeneration.
Unterschied zwischen Amyloid und Prion
Definition und Pathophysiologie
Amyloide sind Aggregate (eine Formation aus vielen zusammengesetzten Teilen) einer stabähnlichen Struktur, die Wiederholungen einer bestimmten Art von Protein enthält. Wenn abnormale Plasmazellen, die aus den Knochenmarke stammen. Diese Ablagerungen können in jedem lebenswichtigen Organ im Körper (wie Gehirn, Herz oder Darm) aufgebaut werden, was zu verschiedenen schwerwiegenden Erkrankungen führt.
Prionen sind eine abnormale Form oder Faltung der spezifischen Proteine von Amyloiden im Gehirn, wodurch sie ansteckend und in der Lage sind, unbegrenzt zu erneuern. Mit anderen Worten, Prionen werden als Unterklasse von Amyloiden definiert, in denen die Proteinaggregation infektiös geworden ist und den Zustand der Selbstproduktion verändert hat. Die Prionen haben die Fähigkeit, ihre fehlgefaltete Proteinform in dieselben Proteine zu übertragen, die in einem normalen Zustand auftreten.
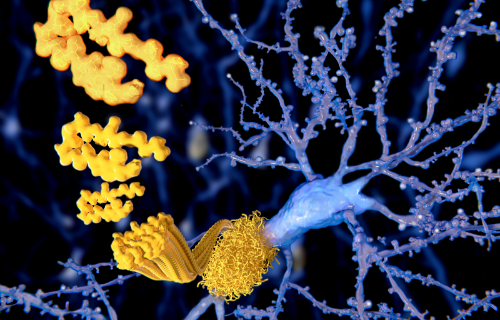
Prionen sind für die neurodegenerativen Störungen verantwortlich, die beim Menschen und Tieren auftreten können. Dies ist darauf zurückzuführen, dass das missbilligte Protein schwieriger durch Enzyme abbaut und sich stattdessen in den Neuronen ansammelt, was zu Zerstörung führt. Wenn diese Neuronzerstörung fortschreitet, veranlasst sie allmählich, dass das Gewebe des Gehirns ein mit Löchern gefülltes schwammartiges Muster aufweist.
Mögliche Ursachen
Amyloide treten auf, wenn das Knochenmark abnormale Blutplasmazellen erzeugt. Diese abnormalen Plasmazellen bilden dann abnormale Arten von leichten Kettenproteinen. Wenn die abnormalen leichten Kettenproteine in das Zirkulationssystem (Blutstrom) eintreten, lagern sie sich in wichtige Organe im gesamten Körper ab. Dies kann an:
- Genetische oder erbliche Komponenten (die die Augen, das Herz, die Nieren und das Gehirn beeinflussen)
- Chronische Krankheiten und Krebsarten
- Zellmutationen
- Seltene Infektionen
Prionen sind die missbilligten Proteine, die am Gehirn abgelagerte Amyloide verursachen. Genetisch ist das PRNP -Gen dafür verantwortlich, den Körper auf Produkt -Prionprotein zu lenken. Wenn dieses Gen mutiert, kann es eine abnormale Proteinproduktion verursachen, was zu spezifischen Prionerkrankungen des Gehirns führt. Prionerkrankung kann auch sporadisch auftreten, was zu anderen spezifischen Krankheiten des Gehirns führt.
Symptome
Die Symptome abnormaler Amyloidproteinablagerungen variieren und hängen davon ab, welches Gewebe oder Organ von den Ablagerungen beeinflusst wurde.
Prionerkrankung, verursacht durch missbilligte Prionproteine, die zu Amyloiden im Gehirn führen, zeigt häufig die folgenden Symptome:
- Gedächtnisschwierigkeiten und Änderungen im Urteilsvermögen
- Persönlichkeitsänderungen
- Verwirrung, Schwierigkeit mit Sprache und/oder Desorientierung
- Änderungen in der Koordination und unfreiwilligen Muskelkrämpfen
- Verringerte Qualität des Sehens oder Blindheit
Diagnose
Die Diagnose des Auftretens von Amyloiden in den betroffenen Organen kann speziell durch Biomarker -Tests durchgeführt werden. Dazu gehören die Messung der Änderungen der Größe des Organs und seiner Funktionen, der Messung der Spiegel spezifischer Proteine im Blut, in der Cerebrospinalflüssigkeit und bei Scans (MRT, CT, PET).
Die Diagnose von Prionproteinen im Gehirn, die Prionerkrankungen verursachen, kann durch Scans des Gehirns durchgeführt werden.
Behandlung
Die Behandlung von Amyloiden hängt davon ab, wo sie auftreten. Die einzige Möglichkeit, Amyloide, die produziert zu werden. Es gibt keine andere Möglichkeit, Amyloide zu behandeln, und die durch das Auftreten von ihnen verursachten Störungen werden auf einer bestimmten Basis behandelt.
Prionen, die Prionerkrankungen verursachen, können mit Medikamenten, unterstütztem Leben in Verbindung mit Neurodegeneration, Aufrechterhaltung der Hydratation und Nährstoffaufnahme behandelt werden. Dies heilt nicht die Produktion von Prionen oder Neurodegeneration, sondern bietet eine Möglichkeit zur Verbesserung der Lebensqualität und zur Behandlung der damit verbundenen Symptome.
Vergleichsvergleich zwischen Amyloid und Prion
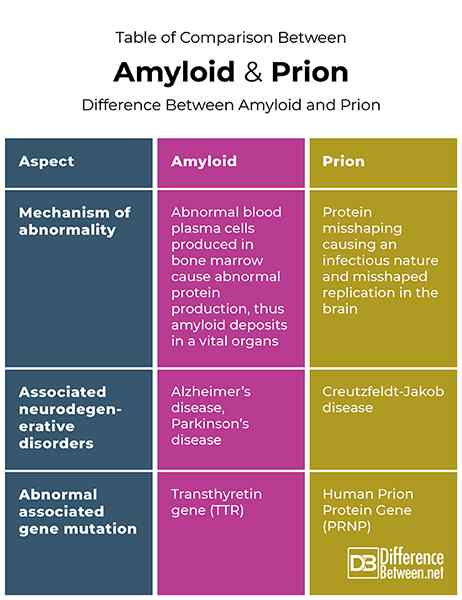
Zusammenfassung von Amyloid gegen Prion
Amyloide und Prionen sind beide mit Protein missbräuchlicher und bestimmter neurodegenerativer Störungen assoziiert. Im Fall von Amyloiden können missbilligte Proteine in jedem Organ Störungen verursachen, an denen sie abgelagert werden. Prionen sind jedoch die missbilligten Proteine, die Amyloide im Gehirn verursachen und auf das Gehirn beschränkte Störungen aufweisen können. In Bezug auf neurodegenerative Erkrankungen sind Amyloide für die Alzheimer-Krankheit und die Parkinson-Krankheit verantwortlich, bei denen Prionen für die Creutzfeldt-Jakob-Krankheit verantwortlich sind.
FAQ
Sind Prionen eine Art Amyloid?
Amyloide sind Aggregate mit einer fibrillären Struktur, die aus Wiederholungen eines bestimmten Proteins bestehen. Prionen werden als Unterklasse von Amyloiden definiert. Bei Prionen wird die Proteinaggregation selbst selbstverpackend und dann infektiös-dies ist die Ursache für viele tödliche neurodegenerative Erkrankungen wie die Creutzfeldt-Jakob-Krankheit.
Wie hängen Amyloidplaques mit Prionen zusammen??
Amyloid-Plaques wurden im Gehirn von Menschen mit Creutzfeldt-Jacob-Erkrankung gefunden. Creutzfeldt-Jacob-Krankheit ist eine durch Prionen verursachte Krankheit. Scrapie ist eine weitere Prionerkrankung, diesmal bei Tieren, mit Amyloidplaques, die aus Prionproteinen bestehen
Was ist der Unterschied zwischen einem Prion und einem Protein?
Der Hauptunterschied zwischen einem Protein und einem Prion liegt in der Struktur - Prionen werden fehlgefaltet, was dazu führt.
Ist Amyloid Beta ein Prion?
Amyloid Beta teilt aufgrund seiner Fähigkeit, sowohl selbst zu propagieren als auch die Existenz mehrerer verschiedener „Stämme“, viele ununterscheidbare Eigenschaften mit Prionen. Diese Eigenschaften machen es wahrscheinlich, während Krankheiten ein Prion zu werden.
- « Unterschied zwischen Reizdarmsyndrom und Lactose -Intoleranz
- Unterschied zwischen elektromotiver Kraft und Magnetomotivkraft »